

What is coverage
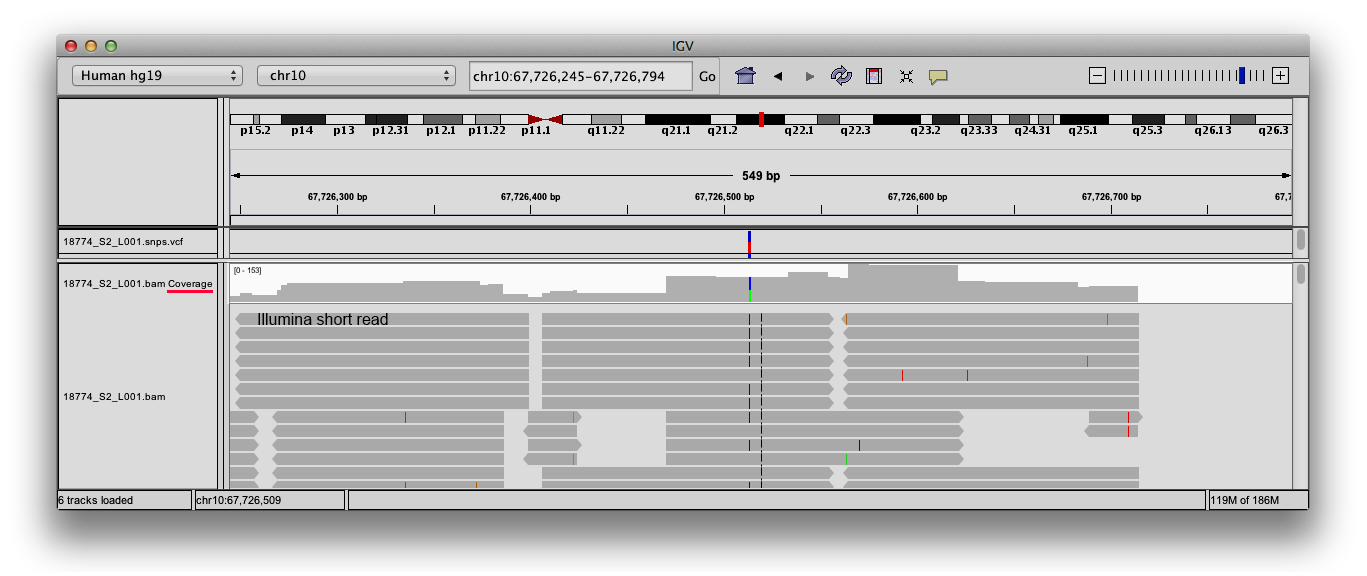
Sequence coverage (or sequencing depth) refers to the number of reads that include a specific nucleotide of a reference genome. In the screenshot below a small region of the Human genome is shown in a genome browser, where the alignment of a re-sequencing experiment was loaded. Each gray arrow in the main area of the window represents a single read, while the highlighted area is a coverage plot, summarizing for each nucleotide how many times was sequenced in this experiment.
If the sequencing was performed using paired ends or mate pairs, it’s possible to physical coverage is the number of times a base is read or spanned by paired ends or mate paired reads¹. The picture below shows the difference.
Bedtools genome coverage
Bedtools has an even better option to calculate coverage, as it produce a standard BED file.
bedtools genomecov -bga FILE.bam > FILE.coverage.bed
The output is a standard BED file: a tabular file with these columns: chromosome, start, end, coverage. The difference with the previous output is that adjacent bases with the same coverage will be collapsed in the same interval.
If we want to quickly detect the zero coverage regions, we can exploit the fact that the last column indicates the coverage:
bedtools genomecov -bga FILE.bam | grep -w "0$" > FILE.0X.bed
Both samtools and bedtools can extract coverage information from a BAM file. Using text manipulation tools (like grep or awk) we can further filter the output they produce to select regions of interest.
Getting the regions covered within a coverage range

A quick tool to produce a coverage profile is covtobed, that takes a sorted BAM file as input and will produce a BED file, optionally specifying to only prints those intervals having a coverage within a range (or only the regions with no coverage).
To install the tool you can use Miniconda:
conda install -y -c bioconda covtobed
An then the usage is as simple as:
covtobed -m 0 -M 1 input.bam > not_covered.bed
the full manual is available in the repository wiki.
 pdftotext is a command line tool for converting
pdftotext is a command line tool for converting 
You must be logged in to post a comment.